分子能源学模拟的先容
分子能源学模拟(Molecular Dynamics Simulation, MD)是一种基于经典力学或量子力学旨趣的策画规律,通过数值求解粒子系统的融会方程来商讨分子体系的动态行为。
自1950年代由Alder和Wainwright初度引入硬球模子商讨液体行为以来,其应用已膨大到卵白质折叠、材料联想、化学反馈机理以及生物分子互相作用等领域。
分子能源学模拟的中枢方针是通过生因素子轨迹,揭示微不雅结构与宏不雅性质之间的筹商,并弥补实际难以径直不雅测的时空轨范缺口。
举例,在生物医学领域,MD模拟可商讨酶催化机制或药物与靶方向献媚摆脱能;在材料科学中,它用于展望纳米材料性能或界面扩散行为。
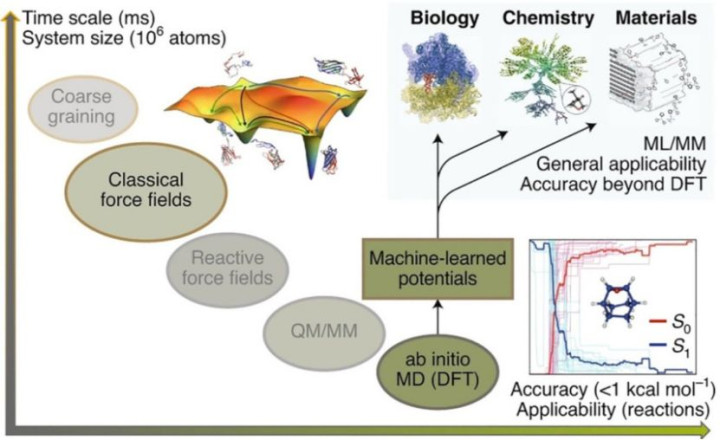
(DOI: 10.1039/d2nr02640f)
凭据表面基础的互异,分子能源学模拟主要分为两类:第一性旨趣分子能源学(Ab Initio Molecular Dynamics, AIMD)和经典分子能源学(Classical Molecular Dynamics, CMD)。
AIMD基于量子力学旨趣,径直通过密度泛函表面(DFT)或波函数规律策画电子结构,从而推导原子间互相作用势能。
举例,Dawson等(2018)使用AIMD模拟水的氢键辘集,发现其能精确捕捉Kohn-Sham能量与氢键动态的关联性,但需均衡高策画本钱与统计可靠性。
CMD则依赖劝诫力场(如CHARMM、AMBER或OPLS)描画原子间作使劲,通过参数化方程简化策画。
举例,GROMACS软件基于牛顿融会方程,通过积分算法生成卵白质轨迹,适用于大范围系统的万古辰模拟。
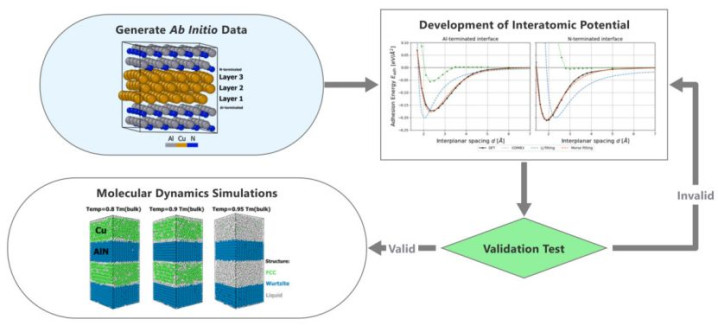
(https://doi.org/10.1021/acsami.2c01347)
第一性旨趣分子能源学模拟(AIMD)的界说与中枢规律
第一性旨趣分子能源学模拟(Ab initio Molecular Dynamics, AIMD)是一种基于量子力学的策画规律,用于商讨分子的融会学和能源学行为。
其中枢在于径直从量子力学的基本方程启程,通过电子结构策画生成势能面,从而模拟原子间的互相作用和系统的演化历程。
这种规律不依赖于劝诫参数,而是通过精确策画电子结构来得到势能、力和能量等物理量,因此梗概提供高度精确的模拟收尾。
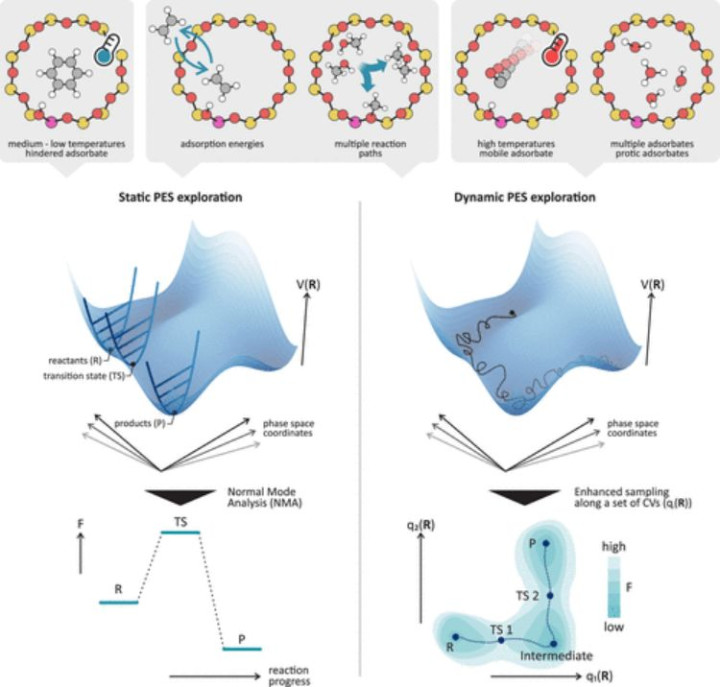
(https://doi.org/10.1021/acscatal.3c01945)
AIMD的中枢规律包括以下几个才气:
1.第一性旨趣电子结构策画:利用密度泛函表面(DFT)或其他量子力学规律策画系统的基态电子结构,包括电子密度、能量以及电子-离子互相作用等。
2.势能函数构建:基于电子结构策画收尾,生成描画原子间互相作用的势能函数。这些势能函数频繁包括交换关联能、电子-离子互相作用等。
3.牛顿融会方程积分:通过积分牛顿融会方程,献媚势能函数,模拟原子在势能面中的融会轨迹。
4.温度和压力戒指:通过恒温恒压(NPT)等热力学系综规律,戒指模拟历程中的温度和压力,以商讨系统在不同要求下的行为。
AIMD的上风在于其高精度和可靠性,但其策画本钱较高,尤其是在处理大范围系统时。因此,商讨者频繁献媚增强采样时刻(如摆脱能采样)来提高成果,并通过并行策画优化大范围模拟。
第一性旨趣分子能源学模拟是一种基于量子力学的先进策画规律,豪爽应用于化学、材料科学等领域,用于商讨分子结构、反馈机理以及材料性能等复杂问题。
经典分子能源学模拟(MD)的界说与中枢规律
经典分子能源学(Classical Molecular Dynamics, MD)模拟是一种基于经典力学的策画规律,用于商讨多原子系统在时辰演化中的行为。其中枢规律是通过求解牛顿融会方程,描画系统中粒子的互相作用和融会轨迹。
经典MD模拟频繁使用劝诫势函数(如Lennard-Jones势和库仑势)来描画粒子间的互相作用,这些势函数梗概模拟分子间键合、非键合互相作用以及长程力(如静电作用)。
(DOI: 10.1039/d2nr02640f)
经典MD模拟的基本才气:
1.运行化系统:设定系统的运行树立,包括粒子的位置、速率和加快度。
2.积分牛顿融会方程:通过数值积分规律(如Verlet算法)策画粒子鄙人一时辰步的位置和速率。
3.重迭策画:迭代上述历程,直到达到所需的时辰步长或罢手要求。
4.分析收尾:从轨迹中索求统计量,如结构因子、扩散整个等。
经典MD模拟的特质:
适用范围广:适用于从纳米轨范到宏不雅轨范的多种系统,如固体、液体和顺体。
策画本钱相对较低:相较于量子力学规律,经典MD模拟对硬件要求较低,允洽大范围系统的万古辰模拟。
简化假定:忽略电子摆脱度,仅议论原子核的融会,ag百家乐大平台这使得策画愈加高效。
但是,经典MD模拟也存在局限性:
无法处理量子效应:关于波及电子行为的首要表象(如化学反馈),经典MD模拟无法提供精确描画。
时辰轨范戒指:由于需要积分牛顿方程,经典MD模拟的时辰步长受限于系统的频率,频繁只可模拟毫秒级的时辰范围。
经典MD模拟的中枢在于通过经典力学和劝诫势函数,献媚数值积分规律,对多原子系统的动态行为进行展望和分析。这种规律在材料科学、生死亡学和物理学等领域具有豪爽应用。
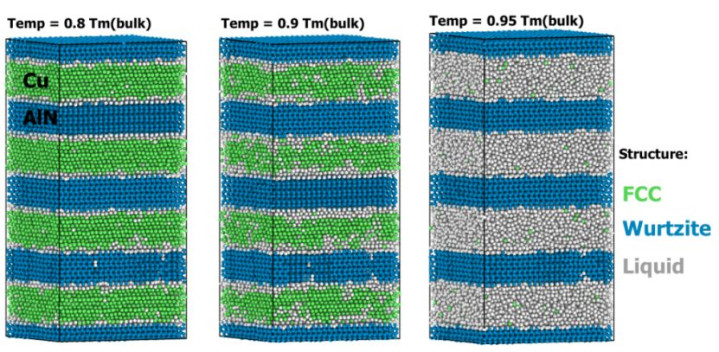
(DOI: 10.1039/d2nr02640f)
AIMD和MD的优污点
两者的优污点对比在多个文件中得到潜入探讨。
AIMD的上风在于高精度:举例,Liu等(2022)指出,AIMD能准确展望非晶硅和硫族化合物玻璃的原子胪列,而经典力场因忽略电子极化效应可能导致盐桥作用被高估。
此外,AIMD在催化反馈旅途商讨中不成或缺,如沸石催化中动态活性位点的识别需依赖量子力学势能面。
但是,AIMD的策画本钱极高,Dawson的模拟透露,1.85纳秒的水分子轨迹需32个并行模拟,总耗时远超经典规律。这戒指了其系统范围(频繁
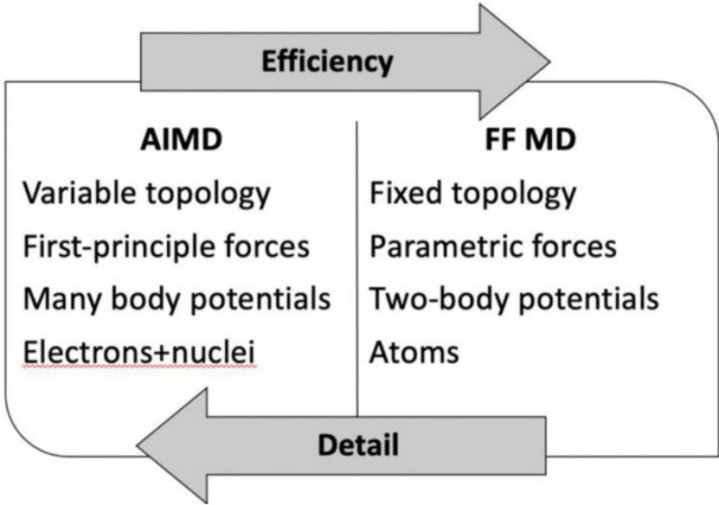
比拟之下,CMD通过劝诫力场罢了高效策画。举例,熔融淬火法模拟玻璃酿成时,CMD可处理百万原子级系统,且与实际数据吻合邃密。
其局限性在于力场参数化依赖特定体系:如Campetella(2015)比较AIMD与CMD对离子液体氢键的模拟,发现经典力场虽能复现合座结构,但无法精确描画羧酸酯氧原子的动态波动。
此外,CMD对化学反馈或电子窜改历程的描画能力有限,需献媚量子力学/分子力学(QM/MM)羼杂规律。
举例,在酶催化商讨中,QM/MM可对活性位点进行量子处理,而周围环境秉承经典力场,均衡精度与成果。
应用场景的采选需量度精度与策画资源。
在生物大分子(如卵白质折叠)或材料界面扩散商讨中,CMD因能模拟微秒级轨迹而更具上风;而在波及电子结构变化(如催化剂联想或金属-有机框架)时,AIMD不成替代。
连年来,机器学习势能(MLPs)的兴起为两者架设桥梁:通过熟习量子力学数据生成高遵守场,MLPs既能保抓AIMD精度,又膨大了CMD的轨范。
举例,Deepmd-kit在Li10GeP2S12固态电解质中的应用,通过交融第一性旨趣与经典规律,显耀擢升了离子扩散模拟的可靠性。
(doi:10.1088/1742-6596/2713/1/012071)
综上,分子能源学模拟作为跨学科器具,其分类与采选需献媚具体科常识题。AIMD与CMD的互补性在复杂体系商讨中尤为凸起,而时刻跳跃(如增强采样、粗粒化和羼杂规律)将抓续拓展其应用鸿沟。
找华四肢念策画👍专科靠谱宽心又省时!
益于表面策画化学的快速发展,策画模拟在纳米材料商讨中的利用日益豪爽而潜入。科研领域还是渐渐酿成了“精确制备-表面模拟-先进表征”的商讨形态,而恰是这种实际和策画模拟的荟萃佐证,愈加增添了论文的可靠性和严谨性,频频梗概得到更豪爽的招供。
“实际+策画”的形态已渐渐成为顶刊标配!华算科技是专科的表面策画与科研测试惩办决策就业商,为高校和企业的科研团队提供材料、催化、能源、生物等领域的表面策画和测试表征惩办决策。
华算科技已向国表里1000多家高校/科研单元提供了独特50000项表面策画和测试表征就业下载AG百家乐,部分策画数据已发表在Nature & Science正刊及大子刊、JACS、Angew、PNAS、AM系列等外洋顶刊。
下一篇:没有了